At the atomic level, the world is a symphony of interactions where positively charged nuclei are encircled by tempestuous negatively charged electrons. The complexity of atomic behavior intensifies when these individual atoms coalesce to form molecules. As they engage with each other, the electrons of these atoms interact in an intricate dance that represents one of the quintessential challenges in modern science: simulating these interactions accurately and efficiently. Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin, in collaboration with the tech giant Google DeepMind, have embarked on a groundbreaking journey to transcend the limitations of traditional computation in this domain through innovative machine learning (ML) algorithms.
The Challenges of Traditional Methods
For decades, scientists have relied on the Schrödinger equation to decode the energy configurations of quantum systems. This foundational equation lays the groundwork for understanding atomic and molecular behavior but achieving numerical solutions becomes an Everest to scale as the molecule’s size increases. The scenario is dire—solving this equation can take considerable time, often stretching into days on powerful computers, particularly when examining complex molecules containing dozens of atoms. This reality poses a formidable barrier for researchers aiming to study dynamic chemical processes over extensive time frames, as the computation necessary can skyrocket, utilizing resources beyond what current technology can offer.
The painstaking task of modeling molecular dynamics—integral for the advancement of materials science and drug discovery—often hinges on the ability to accurately predict interactions that dictate everything from protein folding to biomolecular binding. As highlighted by Thorben Frank from BIFOLD, the capacity to simulate these dynamics could lead to immense efficiencies, potentially averting the labor-intensive and costly experimental methods that currently dominate the field.
Machine Learning: A Beacon of Hope
Fortunately, the landscape of molecular simulation is witnessing a transformative shift thanks to the advent of machine learning technologies. Unlike traditional approaches that rely on the explicit solving of mathematical equations, ML models can learn trend patterns in atomistic interactions. They require far fewer resources and time while providing predictions about electronic interactions at a granular level. This shift effectively redefines the computational paradigm and holds enormous potential to streamline the discovery of materials and drugs.
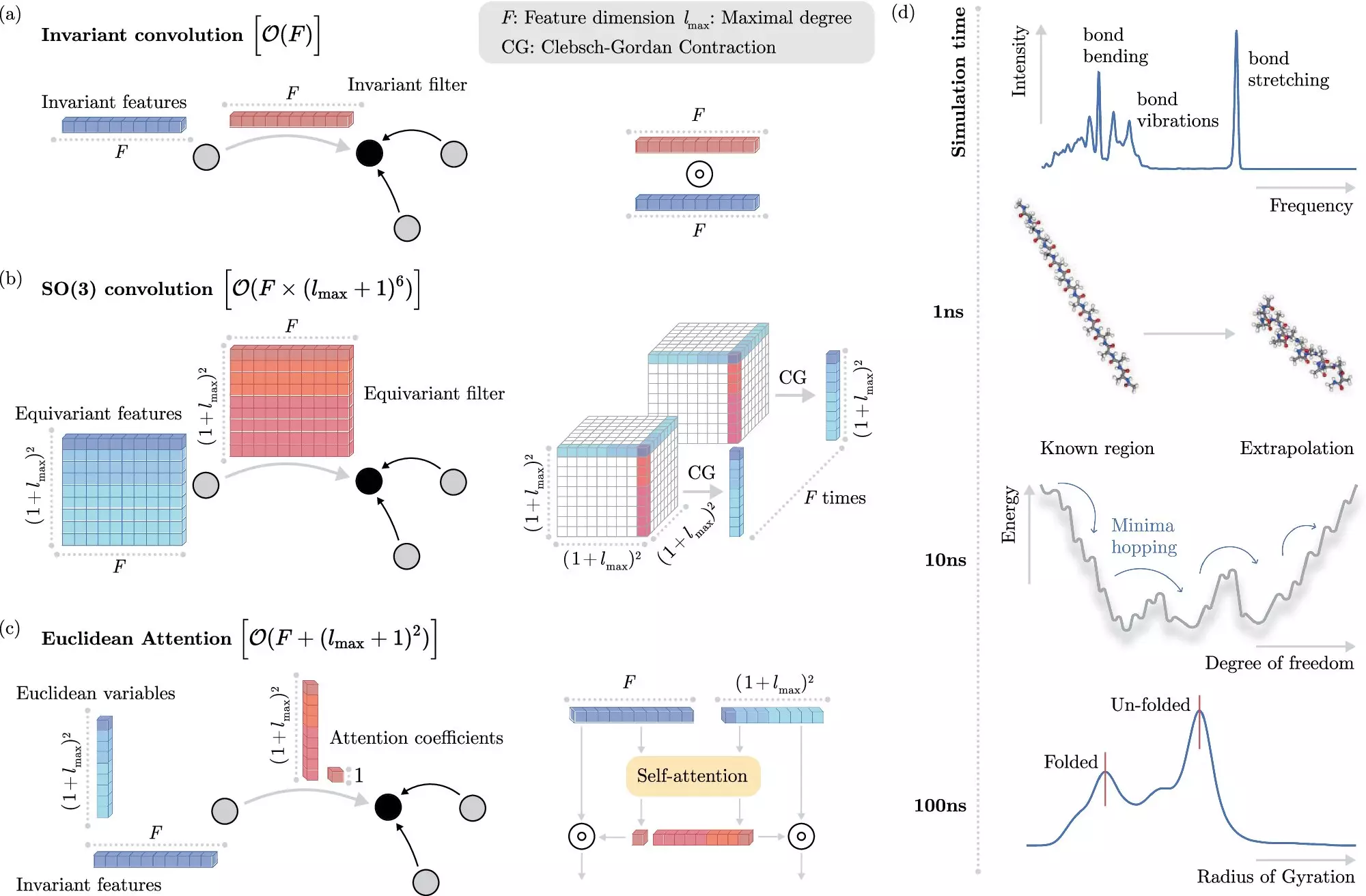
The challenge, however, lies not only in creating robust machine learning models but also in teaching them how electrons interact without being hampered by complexity. Many algorithms attempt to leverage fundamental physical principles called “invariances,” which maintain certain characteristics of molecules regardless of their spatial arrangement. Yet, incorporating these invariances into machine learning models often comes at a high computational cost, hampering speed and efficiency.
The Breakthrough Algorithm
Addressing these limitations, the researchers at BIFOLD have introduced an avant-garde learning algorithm that elegantly decouples the task of invariance from other chemical system characteristics. This novel approach eliminates the computational gridlock experienced in previous models, allowing the ML system to focus on more pivotal physical information. As a result, simulations that once required formidable clusters of high-performance computers for months can now be executed on a single node in just days.
The implications are profound: the ability to conduct long-term molecular simulations opens up new avenues for comprehensive analysis of chemical systems, presenting an opportunity for richer insights into foundational natural phenomena. As Dr. Stefan Chmiela notes, this leap in efficiency marks a critical turning point that can fundamentally alter our perception and understanding of the atomic world.
Towards Practical Applications
The potential for real-world applications stemming from these advanced simulations is staggering. Consider, for instance, the promise that revolves around drug development. Machine learning can streamline the identification of stable molecular structures, such as the crucial docosahexaenoic acid (DHA) that serves as a vital building block in human cognition. Traditional methods would have found it nearly impossible to undertake such exhaustive searches involving thousands of candidates, but with this new ML-driven mechanism, previously inconceivable accuracy and efficiency become attainable.
As researchers work alongside advanced machine learning frameworks, they are not only expediting the discovery process but also optimizing it for sustainability by minimizing the need for extensive laboratory experiments. This combination of computational power and physical understanding is seen by experts like Prof. Dr. Klaus-Robert Müller, co-director at BIFOLD, as a watershed moment in computational chemistry that promises to break down barriers long resistant to scientific inquiry.
In a landscape teeming with possibilities, it is becoming increasingly clear that the synthesis of machine learning and quantum chemistry is charting a new course for science. This remarkable synergy holds the potential to illuminate the many intricate threads that tie together the micro and macro dimensions of our universe, guiding humanity toward future innovations that were once relegated to the realm of dreams.
Leave a Reply